Chukwudi Anago-Amanze, MD, MPH1, Sameera Shuaibi, MD1, Dhaval Patel, BS1, Jennifer von Ende, MD2, Jonathan Mizrahi, MD1 1Ochsner Health Clinic Foundation, New Orleans, LA; 2Ochsner Medical Center, New Orleans, LA Introduction: Muir-Torre Syndrome (MTS), a rare autosomal dominant variant of Lynch syndrome, arises from germline mutations in DNA mismatch repair genes, most commonly MLH1 and MSH2¹. It is characterized by sebaceous skin neoplasms and visceral malignancies, particularly colorectal and genitourinary cancers².
Case Description/
Methods: Diagnosed with Stage II colon cancer in 1993, he underwent a hemicolectomy and chemotherapy after years of colonoscopic surveillance. Repeat colonoscopies revealed recurrent advanced adenomas, including high-grade dysplasia in 2023. He later developed prostate cancer, which was treated with pelvic radiation. Eighteen months prior to terminal admission, he experienced intermittent rectal bleeding that progressed to transfusion-dependent hematochezia. Colonoscopy showed rectal angiodysplasias and radiation proctitis, which were treated with argon plasma coagulation and repeat intervention⁴. A mass was discovered in the pancreatic head. A biopsy in 2017 revealed intestinal-type adenocarcinoma with MLH1 loss, while a recurrence in 2025 demonstrated additional PMS2 loss, underlining the clinical significance of repeat immunohistochemical testing as the tumor profile evolves. Imaging indicated retroperitoneal and mediastinal lymphadenopathy, osteolytic vertebral lesions, ascites, and pleural effusions. CA 19-9 levels were elevated. Paracentesis was complicated by persistent oozing in the setting of thrombocytopenia. A non-occlusive femoral DVT was not treated with anticoagulation. He later experienced seizures, raising concerns for CNS spread, and was transitioned to hospice. Discussion: This case illustrates the evolving gastrointestinal burden in MTS; from surveillance to managing multifocal malignancies and treatment-related complications, highlighting the longitudinal role of gastroenterology in hereditary cancer care⁵. Given the autosomal dominant inheritance, cascade genetic screening and counseling of at-risk relatives remain essential for early detection and management⁶. References
1. Ponti G, Ponz de Leon M. Muir-Torre syndrome. Lancet Oncol. 2005;6(12):980-987. doi:10.1016/S1470-2045(05)70473-4
2. Roberts ME, Riegert-Johnson DL, Thomas BC, et al. A clinical scoring system to identify patients with sebaceous neoplasms at risk for the Muir-Torre variant of Lynch syndrome. Genet Med. 2014;16(9):711-716. doi:10.1038/gim.2014.27
3. Vasen HF, Blanco I, Aktan-Collan K, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC)
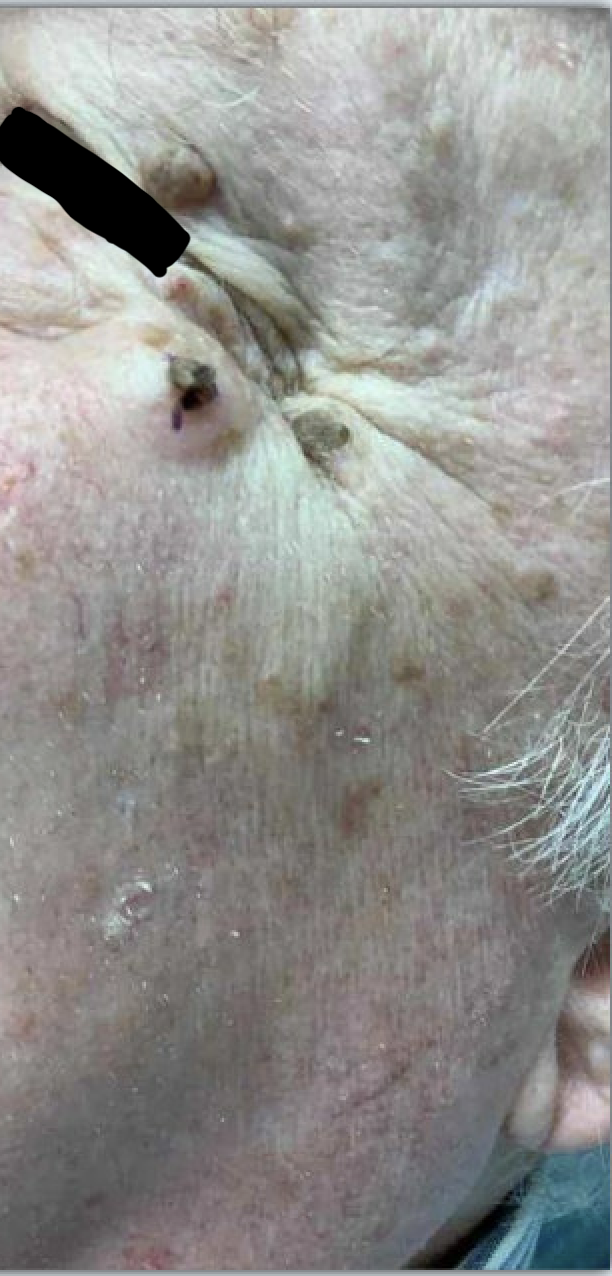
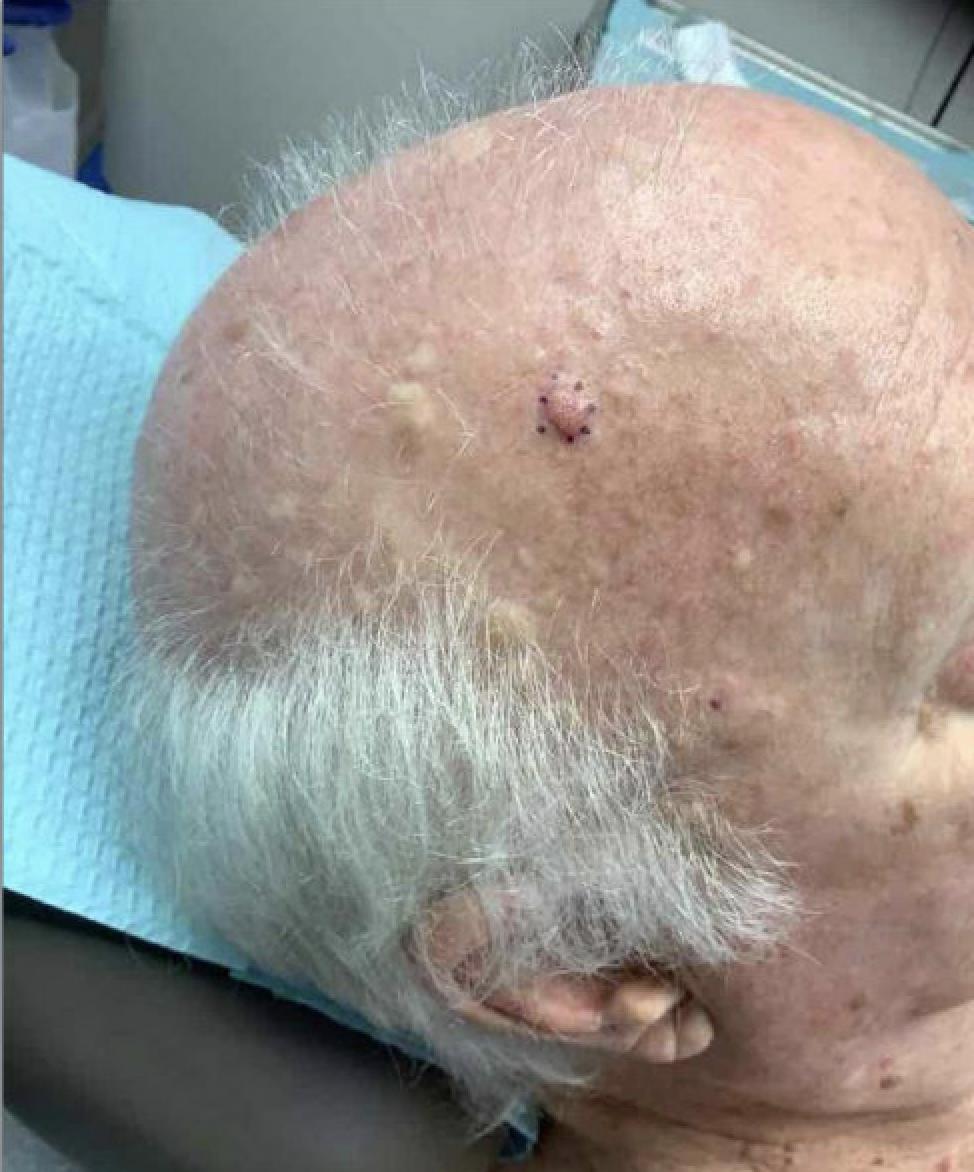
Figure: sebaceous skin lesions involving the face and scalp
Figure: sebaceous skin lesions involving the face and scalp
Disclosures: Chukwudi Anago-Amanze indicated no relevant financial relationships. Sameera Shuaibi indicated no relevant financial relationships. Dhaval Patel indicated no relevant financial relationships. Jennifer von Ende indicated no relevant financial relationships. Jonathan Mizrahi indicated no relevant financial relationships.
Chukwudi Anago-Amanze, MD, MPH1, Sameera Shuaibi, MD1, Dhaval Patel, BS1, Jennifer von Ende, MD2, Jonathan Mizrahi, MD1. P0534 - Gastrointestinal Surveillance and Beyond in Muir-Torre Syndrome: A 30-Year Case Perspective, ACG 2025 Annual Scientific Meeting Abstracts. Phoenix, AZ: American College of Gastroenterology.

 photo")