Stephanie Mrowczynski, BS, MD1, Nicholas Habibian, BS2, Matthew Houle, MD1, Corinne Zalomek, MD1, Ross Bonnot, MD1, Molly Delk, MD1, Husayn F. Ramji, MD2, Anveet Janwadkar, MD1 1Tulane School of Medicine, New Orleans, LA; 2Tulane University School of Medicine, New Orleans, LA Introduction: Bardet-Biedl Syndrome (BBS) is a rare ciliopathic disorder characterized by obesity along with other signs of metabolic syndrome, intellectual disability, retinal dystrophy, polydactyly, cystic kidneys, and hypogonadism. BBS treatment is largely focused on management of metabolic syndrome to prevent various associated complications such as metabolic-dysfunction associated steatohepatitis (MASH), though few cases are documented in the literature. Alpha-1 antitrypsin deficiency is a relatively common disease with highly variable clinical manifestations ranging from minimal biochemical changes to emphysema and liver disease. However, the disease rarely progresses to a degree that necessitates liver transplant.
Case Description/
Methods: A 29y man with a past medical history of BBS, hypertension, diabetes mellitus, and MASH presented to the emergency room for acutely elevated liver function tests on routine outpatient laboratory testing, and two weeks of nausea and vomiting. Physical examination notable for a distended abdomen, bilateral lower extremity edema, jaundice and acanthosis nigricans. Labs significant for AST 130, ALT 41, albumin 3.1, total bilirubin 25.5, INR 1.6, and platelets 92. Abdominal ultrasound and CT of abdomen and pelvis was performed, revealing cirrhosis, ascites, and splenomegaly. MRCP was performed and negative for any obstruction. Liver biopsy revealed PAS-positive inclusions in hepatocytes suggesting alpha-1 antitrypsin (A1AT) deficiency, and genotype analysis confirmed PI*MZ heterozygosity. Ultimately, the patient was listed for transplant and received a deceased donor liver. Discussion: AAT deficiency is inherited in an autosomal co-dominant fashion; homozygotes for the Z allele (PI*ZZ) have severe deficiency of AAT. Heterozygotes are largely asymptomatic, with PI*MS genotype usually producing sufficient amounts of AAT, and PI*MZ having a slight increased risk of developing liver or lung disease.1
Approximately 30% of patients with BBS have hepatobiliary disorders, including liver fibrosis, metabolic dysfunction-associated steatotic liver disease (MASLD), MASH and cystic dilation of bile ducts.2 Review of the current literature revealed no documented cases of BBS-associated liver cirrhosis.
This case uniquely presents a young patient with decompensated cirrhosis, initially attributed solely to MASH secondary to Bardet-Biedl Syndrome who upon further work-up was found to be PI*MZ genotype, illustrating how interacting factors can synergistically lead to need for transplant.
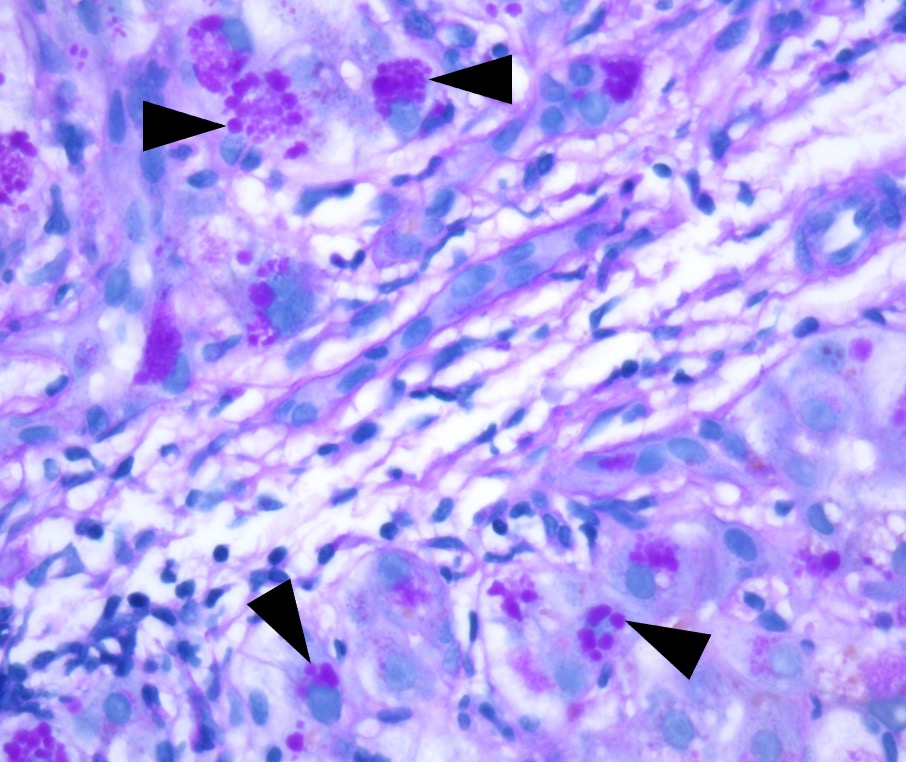
Figure: Figure 1: PAS stain after digestion with diastase highlights intracytoplasmic PAS+ globules (arrowhead) compatible with AAT
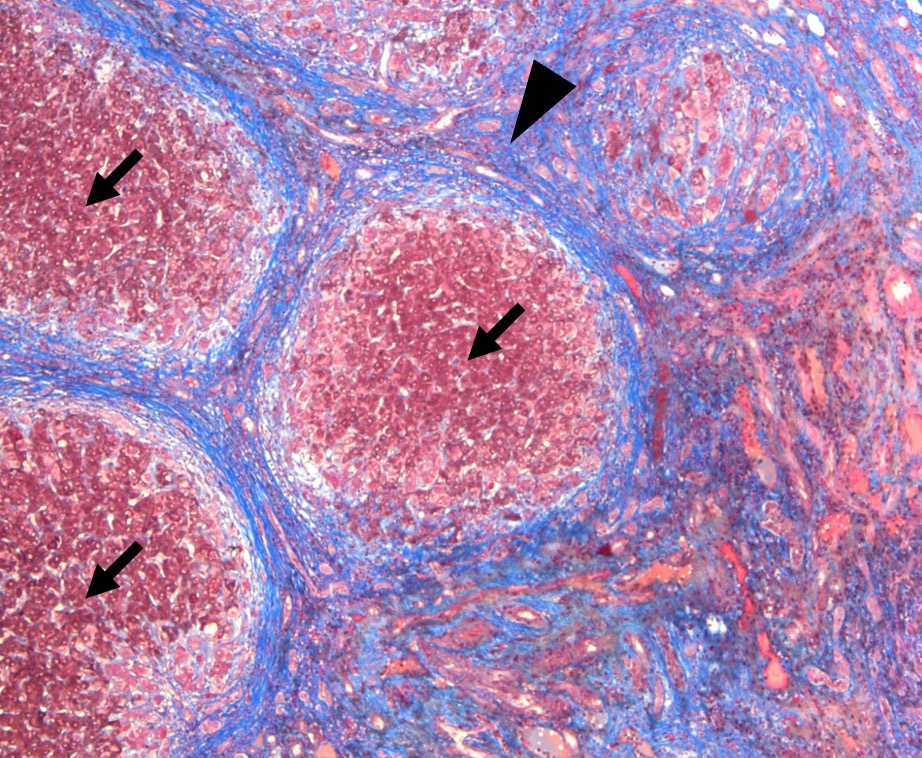
Figure: Figure 2: Trichrome stain highlights regenerative nodules (arrow) surrounded by extensive dense fibrous tissue (arrowhead). These are the microscopic features of cirrhosis.
Disclosures: Stephanie Mrowczynski indicated no relevant financial relationships. Nicholas Habibian indicated no relevant financial relationships. Matthew Houle indicated no relevant financial relationships. Corinne Zalomek indicated no relevant financial relationships. Ross Bonnot indicated no relevant financial relationships. Molly Delk indicated no relevant financial relationships. Husayn Ramji indicated no relevant financial relationships. Anveet Janwadkar indicated no relevant financial relationships.
Stephanie Mrowczynski, BS, MD1, Nicholas Habibian, BS2, Matthew Houle, MD1, Corinne Zalomek, MD1, Ross Bonnot, MD1, Molly Delk, MD1, Husayn F. Ramji, MD2, Anveet Janwadkar, MD1. P4016 - Double Trouble: Liver Transplantation in a Patient With Bardet-Biedl Syndrome and Alpha-1 Antitrypsin Deficiency, ACG 2025 Annual Scientific Meeting Abstracts. Phoenix, AZ: American College of Gastroenterology.