Sebastian Hermida Velasquez, DO1, Natalia Martin, DO2, Thatcher T. Huynh, DO3, Salaah Siddiqui, DO4, Sanskriti Sharma, MD3, Adam Koller, DO4, Karthik Mohan, DO3 1Palmetto General Hospital, Hialeah, FL; 2Larkin Community Hospital, South Miami, FL; 3Larkin Community Hospital, Hialeah, FL; 4Larkin Community Hospital, Miami, FL Introduction: Langerhans cell histiocytosis (LCH) is a rare disease involving immune cell production, more specifically clonal neoplastic deposition of CD1+ dendritic cells. LCH hepatic involvement has been noted to occur in less than 4-5% of all cases. This case highlights the primary manifestation of LCH as acute liver failure and its challenging steps in diagnosis. We illustrate the importance of early histopathological disease confirmation in the setting of an unexplained hepatopathy with pancytopenia.
Case Description/
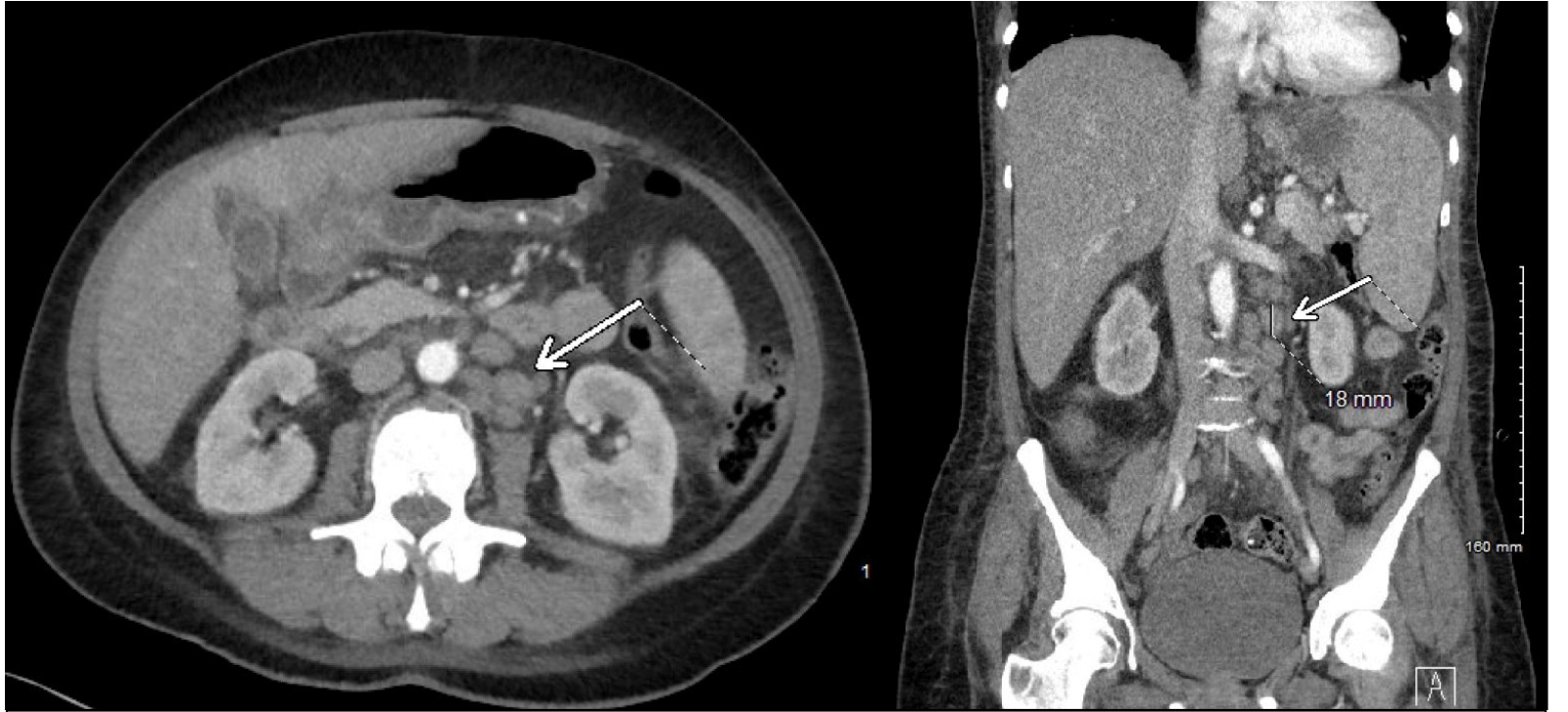
Methods: A 58-year-old female with history of GERD, migraines, and pipeline-embolized aneurysm, presented with 30 lbs of unintentional weight loss, early satiety, and polydipsia. On admission, labs were notable for AST 320 U/L, ALT 280 U/L, total bilirubin 12.4 mg/dL (direct 9.8), INR 2.1, MELD-Na 23, WBC 1.2 K/uL, hemoglobin 7.8 g/dL, and platelets 89 K/uL. MRCP was performed, revealing moderate hepatosplenomegaly with periportal edema, retroperitoneal lymphadenopathy (largest node 2.3 cm) and ascites without any signs of biliary obstruction. Despite negative viral hepatitis serologies, normal ceruloplasmin and autoimmune panel, the patient continued to develop worsening hepatic encephalopathy requiring lactulose regimen. The patient ultimately underwent bone marrow and lymph node biopsies revealing LCH. She was empirically started on high-dose steroids for suspected LCH-induced liver infiltration, stabilized and transferred to a tertiary cancer center for further treatment. Discussion: This case presents LCH in the setting of unexplained liver failure, weight loss and pancytopenia, without any of the typical bone and skin lesions. It is important to note that radiographic absence of biliary duct dilation should not exclude LCH-associated sclerosing cholangitis as it could be possible to identify infiltration with biochemical disturbance prior to any structural changes. Another learning point is the use steroids as a bridge to definitive therapy in rapidly progressive events such as in this patient. Multidisciplinary collaboration was essential in order to navigate the steps required to achieve this diagnosis. LCH’s systemic manifestations, especially in rare presentations can prevent delays in targeted therapy; this case reminds clinicians to remain vigilant for LCH when encountering this constellation of hepatic, hematologic, and constitutional symptoms.
Figure: Figure 1. Contrast-enhanced axial CT abdomen demonstrating hepatosplenomegaly and enlarged retroperitoneal lymph nodes (arrows), largest measuring 2.3 cm in periportal region, with associated mild ascites. Biopsy-confirmed Langerhans cell histiocytosis infiltration.
Disclosures: Sebastian Hermida Velasquez indicated no relevant financial relationships. Natalia Martin indicated no relevant financial relationships. Thatcher Huynh indicated no relevant financial relationships. Salaah Siddiqui indicated no relevant financial relationships. Sanskriti Sharma indicated no relevant financial relationships. Adam Koller indicated no relevant financial relationships. Karthik Mohan indicated no relevant financial relationships.
Sebastian Hermida Velasquez, DO1, Natalia Martin, DO2, Thatcher T. Huynh, DO3, Salaah Siddiqui, DO4, Sanskriti Sharma, MD3, Adam Koller, DO4, Karthik Mohan, DO3. P3977 - Acute Liver Failure as the Primary Manifestation of Langerhans Cell Histiocytosis, ACG 2025 Annual Scientific Meeting Abstracts. Phoenix, AZ: American College of Gastroenterology.