Baylor Scott & White Medical Center Waxahachie, TX
Grace Kim, DO1, Rahul Paryani, MD2, Hema Sadineni, MS2 1Baylor Scott & White Medical Center, Fort Worth, TX; 2Medical Associates of North Texas, Fort Worth, TX Introduction: IgG4-related disease (IgG4-RD) is a fibroinflammatory condition that can affect multiple organs, including the pancreas, kidneys, and hepatobiliary system. It is characterized by elevated serum IgG4 levels, storiform fibrosis, and lymphoplasmacytic infiltrates with IgG4-positive plasma cells. Autoimmune pancreatitis (AIP) and tubulointerstitial nephritis (TIN) are common manifestations of IgG4-RD. However, diagnosis can be challenging when imaging findings and serologic markers do not fully align with established criteria. This case report discusses a patient with chronic pancreatitis, autoimmune features, and IgG4 elevation, raising suspicion for IgG4-RD despite incomplete diagnostic criteria.
Case Description/
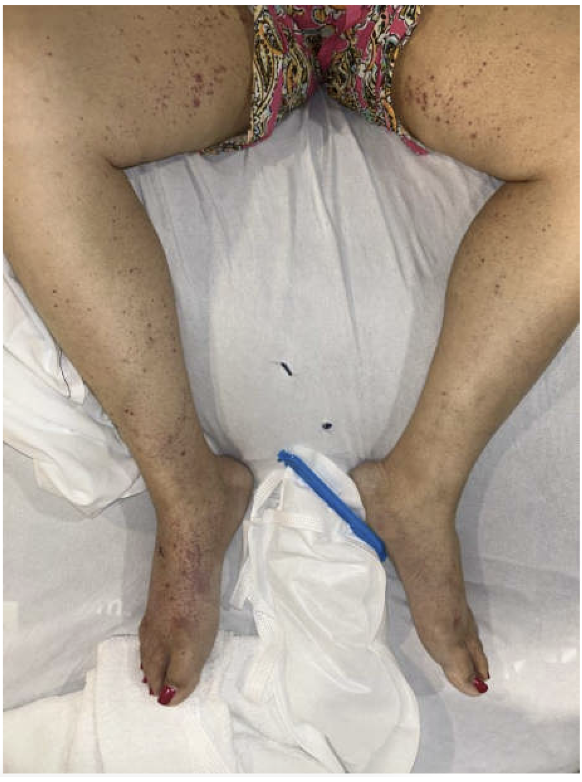
Methods: A 44-year-old female with diabetes, granulomatous infiltration of the right lacrimal gland, and Vogt-Koyanagi-Harada syndrome on adalimumab presented with abdominal pain and hyperbilirubinemia. Endoscopic retrograde cholangiopancreatography revealed a common bile duct stricture requiring stent placement. Her IgG4 level was elevated at 642 mg/dL and cancer antigen 19-9 was negative. Computed tomography showed no clear evidence of AIP but suggested a possible pancreatic head mass. Endoscopic ultrasound (EUS) showed chronic pancreatitis, suspected to be autoimmune in nature, prompting initiation of prednisone therapy. Three years later, imaging again showed a pancreatic head mass. Repeat EUS with biopsy showed stable chronic pancreatitis without biliary abnormalities. Magnetic resonance cholangiopancreatography revealed diffuse pancreatic atrophy without ductal dilation. No evidence of IgG4-related involvement beyond the pancreas was seen. The etiology of her chronic pancreatitis remained uncertain due to incomplete criteria for AIP. Subsequently, she developed progressive renal dysfunction and underwent biopsy, which showed TIN with acute tubular inflammation, patchy interstitial fibrosis (25–30%), and positive p-ANCA. Lab studies revealed elevated erythrocyte sedimentation rate, C-reactive protein, rheumatoid factor, and low complement levels. Skin biopsy of petechial rash showed neutrophilic eccrine hidradenitis. She was treated with high-dose steroids, transitioned to mycophenolate mofetil, and adalimumab was discontinued. Discussion: This case shows the diagnostic challenge of IgG4-RD with multi-organ involvement. Despite lacking classic criteria of IgG4-RD, elevated IgG4, chronic pancreatitis, and TIN supported treatment with steroids and mycophenolate for suspected IgG4-RD.
Figure: Petechial rash over bilateral lower extremities with skin biopsy showing neutrophilic eccrine hidradenitis
Disclosures: Grace Kim indicated no relevant financial relationships. Rahul Paryani indicated no relevant financial relationships. Hema Sadineni indicated no relevant financial relationships.
Grace Kim, DO1, Rahul Paryani, MD2, Hema Sadineni, MS2. P2392 - A Complex Case of IgG4-Related Disease With Multisystem Involvement, ACG 2025 Annual Scientific Meeting Abstracts. Phoenix, AZ: American College of Gastroenterology.