Amrutha Yalla, MD1, Canan D. Dirican, MD2, Sri Lakshmi Ananya Bokka, MD3 1NYMC at St Mary's and Sy Clare's, Denville, NJ; 2NYMC at St Mary's and St Clare's, Denville, NJ; 3Gandhi Medical College & Hospital, Denville, NJ Introduction: Caroli's disease is a rare congenital disorder marked by segmental cystic dilatation of the intrahepatic bile ducts. Patients present with recurrent biliary obstruction, cholangitis, and an increased risk of cholangiocarcinoma. This case highlights the diagnostic and therapeutic challenges in an elderly female with acute cholangitis and a background suggestive of polycystic kidney disease spectrum.
Case Description/
Methods: A 76-year-old woman with a history of hypertension, pancreatic and hepatic cysts, renal cysts, and idiopathic pulmonary fibrosis presented with severe epigastric pain radiating to the back. Patient reported that the pain worsened by food intake and movement. She had a remote history of gallstones and cholecystectomy 30 years ago. Family history was notable for pancreatic, liver, and lung cysts in first-degree relatives. On examination, she was jaundiced with right upper quadrant tenderness but no fever. Laboratory findings revealed cholestatic liver injury with total bilirubin of 6.9 mg/dL, ALT of 644 U/L, ALP of 276 U/L. Tumor markers were elevated with CA 19-9 of 2069 U/mL and CEA of 7.1 ng/mL. Antinuclear antibody was tested considering autoimmune etiology and was negative.
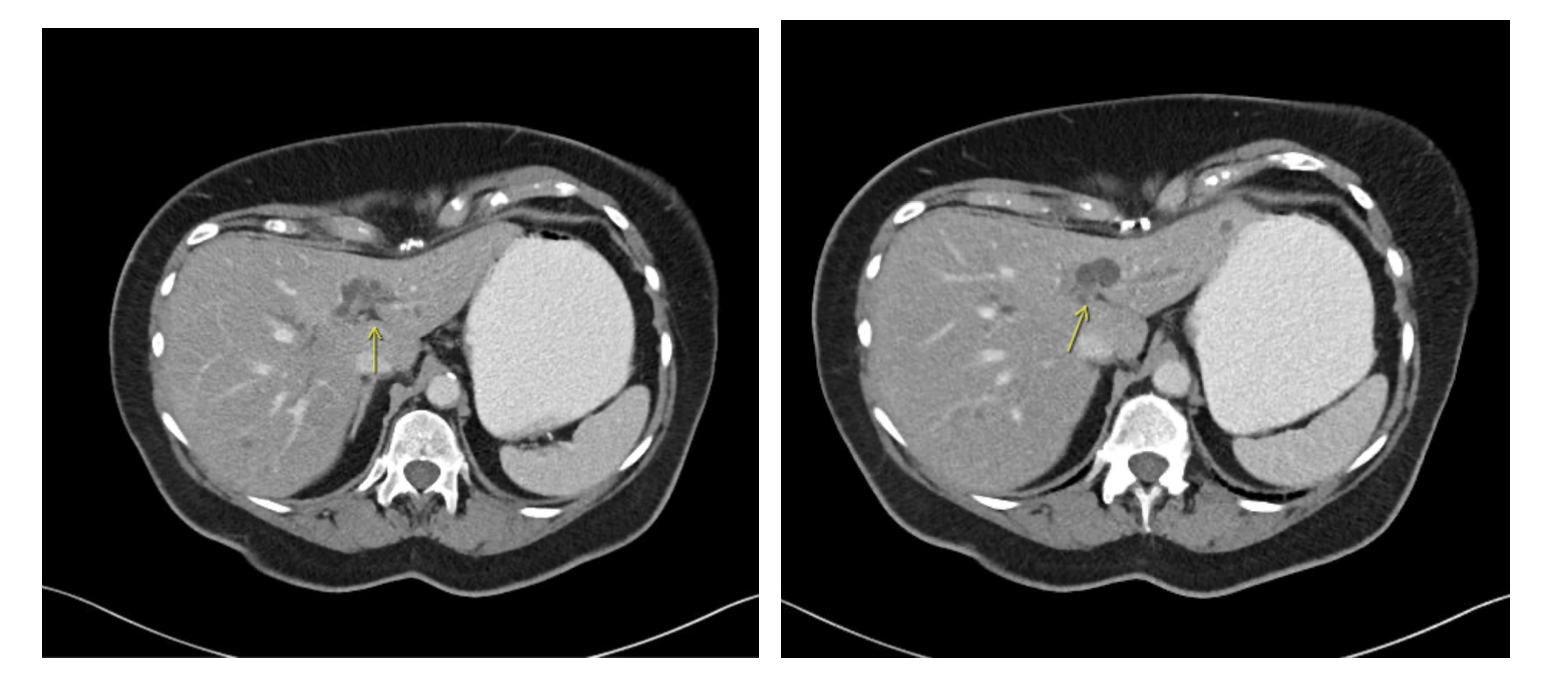
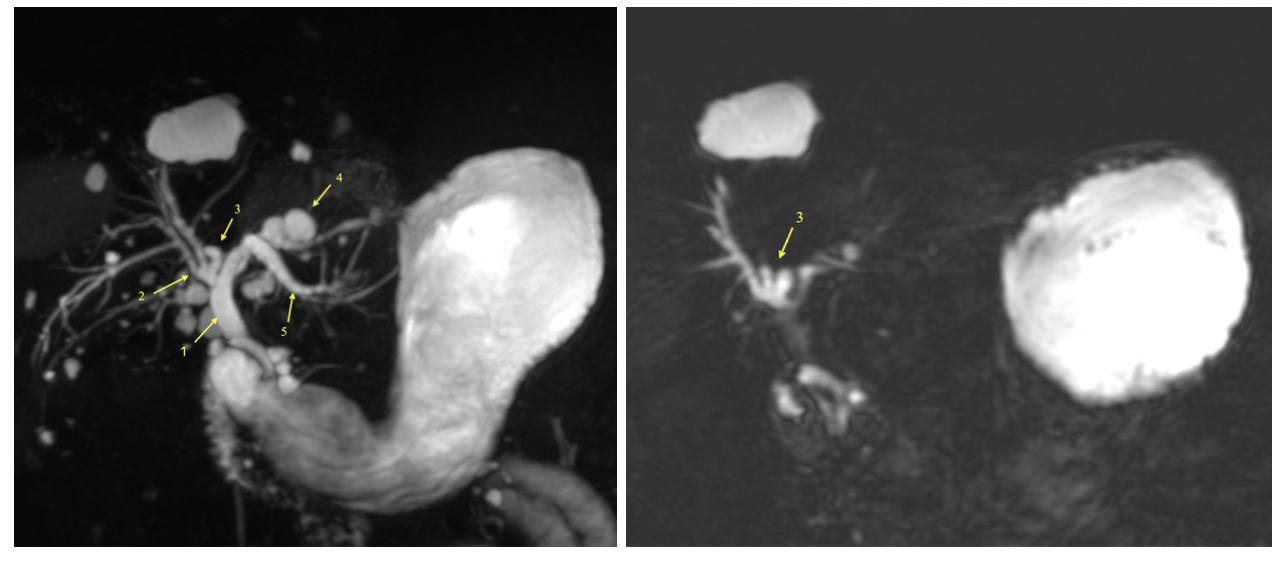
Imaging with CT and MRCP was done and demonstrated intra and extrahepatic biliary duct dilatation, distal common bile duct stones, and multiple hepatic, pancreatic, and renal cysts. ERCP was done and three cholesterol stones were successfully extracted via biliary sphincterotomy and balloon sweeps. Follow-up MRI confirmed the diagnosis of Caroli's disease with characteristic saccular intrahepatic duct dilatations. The patient improved with antibiotics and endoscopic intervention and was discharged with recommendations for regular follow-up. Discussion: Caroli's disease, classified as Type V in the Todani system, is rare with prevalence of less than 1 in 1 million, it may occur solely or as part of the autosomal recessive polycystic kidney disease (ARPKD) spectrum. The clinical presentation is variable, with recurrent abdominal pain, jaundice, and cholangitis as common features. Diagnosis relies on advanced imaging modalities such as MRCP and ERCP. Imaging reveals the pathognomonic segmental cystic biliary dilatations and, occasionally, the "central dot sign". Management is multidisciplinary, involving antibiotics for cholangitis, endoscopic or percutaneous interventions for obstruction, and surgical options for localized disease. Lifelong surveillance is essential due to the risk of cholangiocarcinoma.
Figure: CT scan scan showing cysttic dilations of the intrahepatic bile ducts (showed by yellow arrows)
Figure: MRCP with contrast shows multiple areas of cystic and saccular dilations of the intrahepatic bile ducts.
Disclosures: Amrutha Yalla indicated no relevant financial relationships. Canan Dirican indicated no relevant financial relationships. Sri Lakshmi Ananya Bokka indicated no relevant financial relationships.
Amrutha Yalla, MD1, Canan D. Dirican, MD2, Sri Lakshmi Ananya Bokka, MD3. P1861 - Caroli’s Disease Presenting as Acute Cholangitis: A Rare Case with Polycystic Organ Involvement, ACG 2025 Annual Scientific Meeting Abstracts. Phoenix, AZ: American College of Gastroenterology.

 photo")