Department of Internal Medicine, AdventHealth Orlando Orlando, FL
Award: ACG Presidential Poster Award
Tareq Alsaleh, MD1, Abdul Mohammed, MD2, Medha Karnam, BS3, Torfay Roman, MD2 1Department of Internal Medicine, AdventHealth Orlando, Orlando, FL; 2Department of Gastroenterology and Hepatology, AdventHealth Orlando, Orlando, FL; 3Behavioral and Cognitive Neuroscience, University of Florida, Gainesville, FL Introduction: Erythropoietic protoporphyria (EPP) is a rare disorder of heme synthesis that typically appears in childhood. Here, we present an unusual case of EPP initially presenting as acute liver injury in an adult.
Case Description/
Methods: A 47-year-old female with past medical history of iron-deficiency anemia presented to the emergency department with abdominal pain, jaundice, and vomiting starting three days prior. She denied personal or family history of liver disease, alcohol abuse, or drug use. Vital signs were unremarkable. On physical exam, she had epigastric tenderness without guarding.
Laboratory tests showed significant transaminase, alkaline phosphate, and bilirubin elevations (Table 1). Hepatitis serology was negative. Ultrasound showed a normal gallbladder and hepatic steatosis.
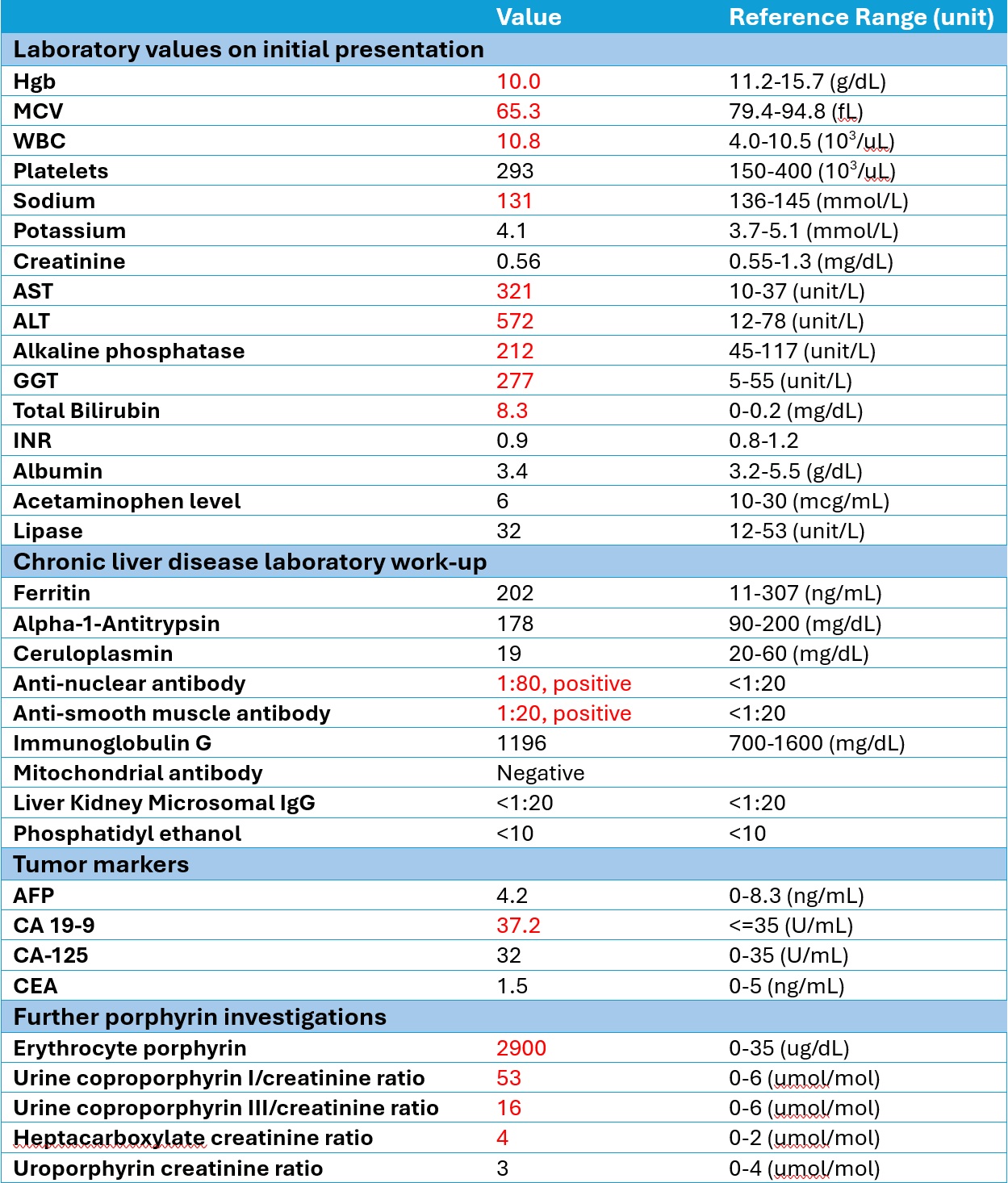
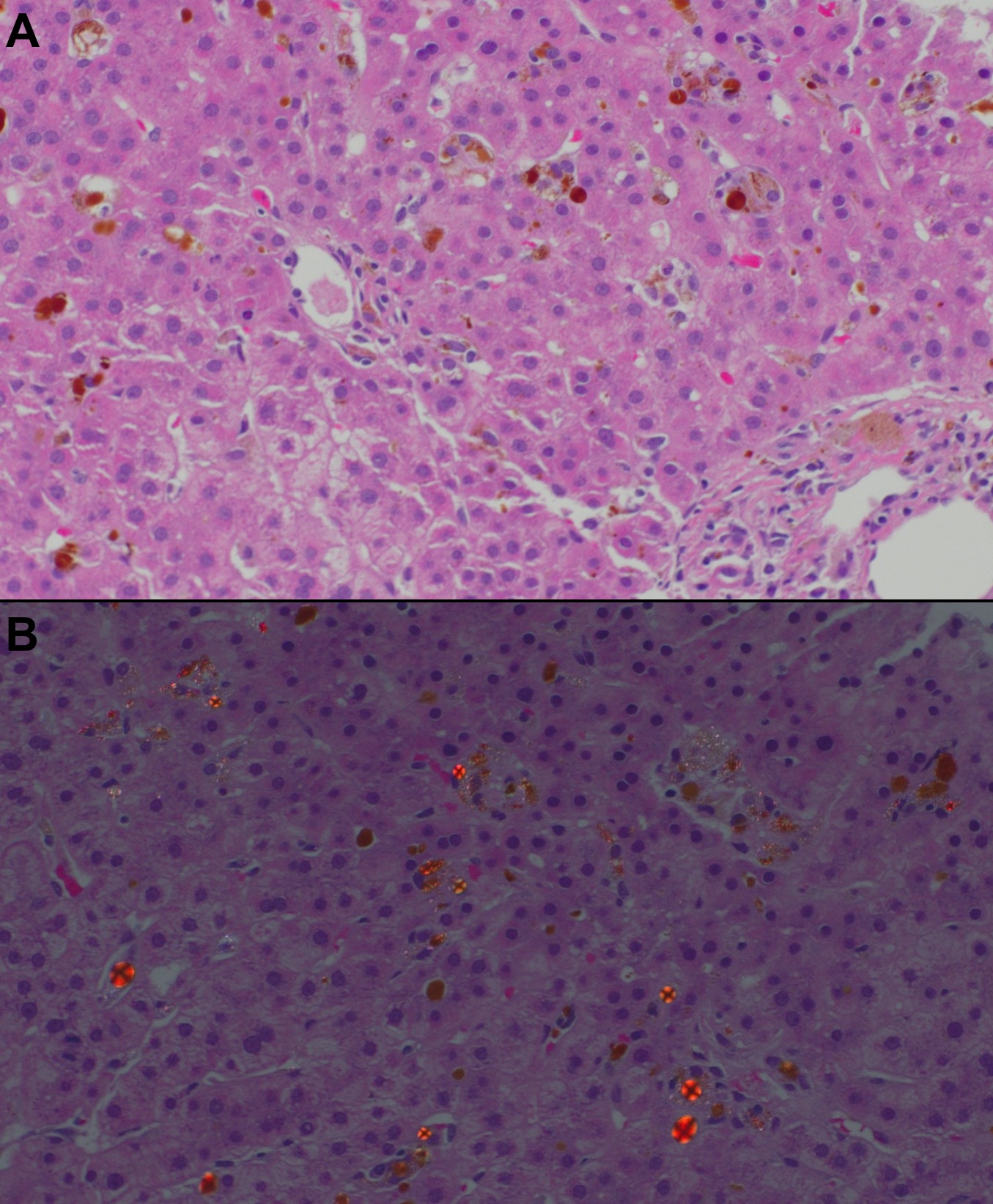
Magnetic resonance imaging of the liver did not report parenchymal or vascular abnormalities. Computed tomography-guided liver biopsy showed severe cholestasis and deposition of Maltese-cross-positive material (Figure 1). Porphyrin blood level was >1000 nmol/L (0-15), with further investigations summarized in Table 1.
She was diagnosed with EPP and treated with IV fluids, analgesics, and anti-emetics. Her symptoms improved and liver chemistry trended down, with the last bilirubin on discharge being 4.7 mg/dL. She was discharged home with an outpatient hematology follow-up for further care. Discussion: EPP is typically due to mutations in ferrochelatase, the enzyme that catalyzes the insertion of ferrous iron into the protoporphyrin IX ring in heme synthesis. This leads to protoporphyrin accumulation in body tissues, including the liver, causing progressive cholestasis and liver damage. Protoporphyric hepatopathy varies from asymptomatic liver enzyme elevations to fulminant acute liver failure. EPP rarely present in adulthood and in most cases, involves the skin before other organs.
Diagnosis is supported by a high erythrocyte protoporphyrin level. Liver biopsy confirms the diagnosis in cases of diagnostic uncertainty. Polarized microscopy shows Maltese-cross patterns of birefringent crystal deposits and plugging of biliary canaliculi with protoporphyrin.
Treatment is targeted towards increasing bile flow, reducing protoporphyrin production, interrupting the enterohepatic circulation and removing the source of protoporphyrin. Options include ursodeoxycholic acid, erythrocyte transfusions, plasmapheresis, cholestyramine, and bone marrow transplant. Severe and rapidly progressive cases require liver transplantation.
Figure: Table 1. Results of laboratory investigations
Figure: Figure 1. A - Canalicular and hepatocytic cholestasis with dense, brown-colored deposit in the canalicular hepatocytes, Kupffer cells, and portal macrophages. B – Polarized microscopy showing Maltese-Cross-positive material.
Disclosures: Tareq Alsaleh indicated no relevant financial relationships. Abdul Mohammed indicated no relevant financial relationships. Medha Karnam indicated no relevant financial relationships. Torfay Roman indicated no relevant financial relationships.
Tareq Alsaleh, MD1, Abdul Mohammed, MD2, Medha Karnam, BS3, Torfay Roman, MD2. P4009 - Acute Protoporphyric Hepatopathy as the Initial Presentation of Erythropoietic Protoporphyria in Adulthood, ACG 2025 Annual Scientific Meeting Abstracts. Phoenix, AZ: American College of Gastroenterology.