Hospital Roosevelt / Gastri-k Guatemala City, San Marcos, Guatemala
Award: ACG Presidential Poster Award
Jorge Pacheco, 1, Ninoska Meléndez, 1, Andres Ruf, 2, Abel Sanchez, MD, MSc3 11. Gastroenterology and Endoscopy Unit, Hospital Roosevelt Guatemala, Guatemala City, Quetzaltenango, Guatemala; 2Hospital Privado de Rosario, Argentina, Rosario, Santa Fe, Argentina; 3Hospital Roosevelt / Gastri-k, Guatemala City, San Marcos, Guatemala Introduction: Progressive Familial Intrahepatic Cholestasis type 2 (PFIC2) is a severe autosomal recessive disorder caused by mutations in ABCB11, leading to dysfunctional bile salt export pump (BSEP) activity and rapid liver disease progression. Benign Recurrent Intrahepatic Cholestasis type 2 (BRIC2) shares genetic overlap but follows an episodic, self-limited course without progressive hepatic injury. This case presents an uncommon disease trajectory marked by recurrent cholestasis with jaundice and pruritus, alternating with asymptomatic phases of persistent biochemical cholestasis, expanding the clinical spectrum of ABCB11-associated disorders beyond conventional classifications
Case Description/
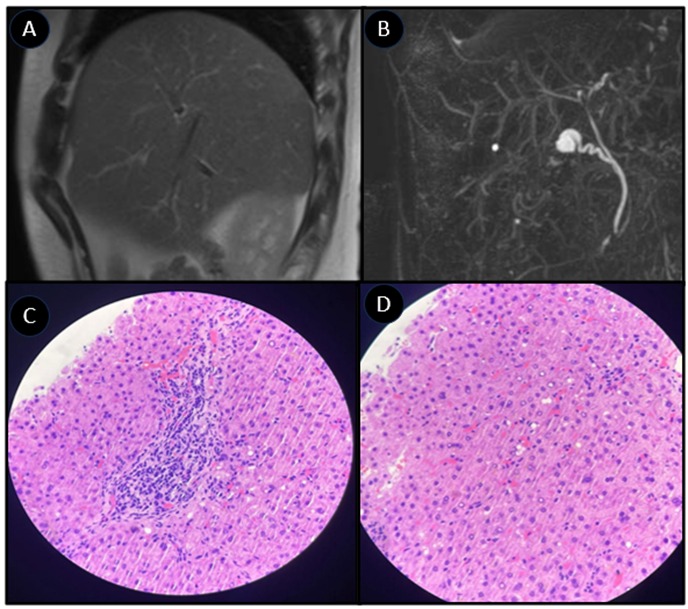
Methods: A 27-year-old male with recurrent jaundice and severe pruritus since age 10 experienced episodes lasting 4–6 months, alternating with persistent biochemical abnormalities despite asymptomatic intervals—including elevated alkaline phosphatase with consistently normal GGT. Imaging ruled out biliary abnormalities, and serological tests excluded autoimmune and infectious causes. During symptomatic episodes, liver biopsy demonstrated lobular necroinflammation, canalicular cholestasis localized to centrilobular zones, and moderate portal and centrilobular fibrosis. In contrast, a biopsy obtained during remission revealed preserved hepatic histoarchitecture, no necrosis, inflammation, or cholestasis, and expansive portal fibrosis with incomplete bridging, alongside perisinusoidal “chicken-wire” fibrosis and pronounced central perivenular fibrosis Genetic testing identified a heterozygous pathogenic ABCB11 mutation (p.Arg470), a variant associated with BRIC2-related BSEP dysfunction, supporting a rare BRIC2 phenotype with atypical chronic hepatic involvement Discussion: This case highlights a rare intersection within ABCB11-associated disorders, demonstrating persistent biochemical cholestasis despite intermittent symptoms, reinforcing the importance of phenotypic variability within BRIC2. The absence of progressive hepatic failure distinguishes this from PFIC2, yet the presence of chronic hepatic fibrosis challenges BRIC2’s traditionally benign course. Whether genetic modifiers or unexplored regulatory mechanisms contribute to this intermediate disease trajectory remains uncertain. This case supports the need for expanded diagnostic frameworks, urging clinicians to reconsider strict disease classifications when evaluating unexplained recurrent cholestasis.
Figure: Figure 1. A, B: Magnetic Resonance Cholangiography" (MRC) demonstrating normal bile ducts anatomy, without any abnormality identified. C, D: preserved hepatic histoarchitecture, presence of expansive portal fibrosis with incomplete bridging, alongside perisinusoidal “chicken-wire” fibrosis and pronounced central perivenular fibrosis.
Figure: Genetic testing identified a heterozygous pathogenic ABCB11 mutation (p.Arg470).
Disclosures: Jorge Pacheco indicated no relevant financial relationships. Ninoska Meléndez indicated no relevant financial relationships. Andres Ruf indicated no relevant financial relationships. Abel Sanchez indicated no relevant financial relationships.
Jorge Pacheco, 1, Ninoska Meléndez, 1, Andres Ruf, 2, Abel Sanchez, MD, MSc3. P1694 - Expanding the ABCB11 Spectrum: Atypical BRIC2 With Persistent Biochemical Cholestasis and Chronic Liver Disease, ACG 2025 Annual Scientific Meeting Abstracts. Phoenix, AZ: American College of Gastroenterology.


 photo")